生成bigwig
By Hongjiang, Feb 27, 2022
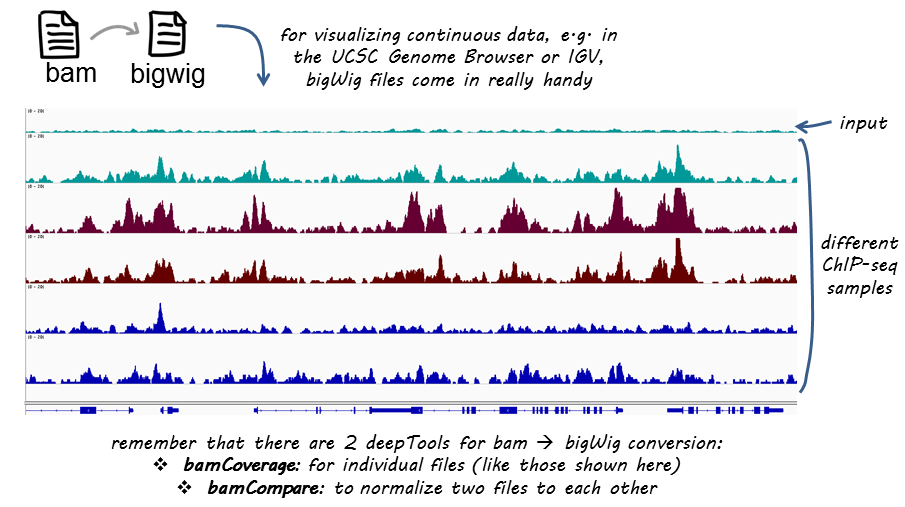
将.bam,.bedGraph等文件转换为 .bigwig文件,缩小文件体积,以便在浏览器中加载。
使用bamCoverage转换
安装Deeptools
一般使用conda安装deeptools。
在使用此程序处理.bam文件前,需要使用samtools index建立索引。
一个最简单的命令为:
必须参数
-b,也可写作--bam,后接输入的.bam文件。
若不指定输出参数,则为stdout,使用 > 来处理。
输出参数
-o,也可写作--outFileName,后接输出的.bigWig文件。
-of,也可以写作--outFileFormat,后接输出文件格式。可选bigwig、bedgraph两种,默认为bigwig。
使用bedGraphToBigWig转换
二进制程序已编译好,可以直接使用。
此程序仅支持Linux使用,UCSC同时提供MacOS版本。
赋予执行权限
下载基因组大小文件
bedGraphToBigWig程序需要基因组大小文件作为参考,使用以下指令下载hg19参考文件。
同理,使用以下指令下载hg38参考文件。
运行程序
bedGraphToBigWig程序需要输入三个变量,第一个是.bedGraph文件地址,第二个是基因组参考文件.chrom.sizes地址,第三个是.bigwig(.bw)文件输出地址。
参考指令:
注:.bedGraph文件需要sort -k1,1 -k2,2n。
注:UCSC也提供bigWigToBedGraph软件。